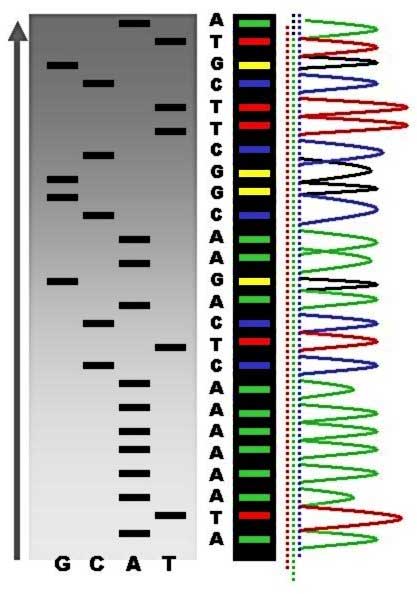
The term DNA sequencing refers to sequencing methods for determining the order of the nucleotide bases—adenine, guanine, cytosine, and thymine—in a molecule of DNA. Knowledge of DNA sequences has become indispensable for basic biological research, other research branches utilizing DNA sequencing, and in numerous applied fields such as diagnostic, biotechnology, forensic biology and biological systematics. The advent of DNA sequencing has significantly accelerated biological research and discovery. The rapid speed of sequencing attained with modern DNA sequencing technology has been instrumental in the sequencing of the human genome, in the Human Genome Project. Related projects, often by scientific collaboration across continents, have generated the complete DNA sequences of many animal, plant, and microbial genomes. The first DNA sequences were obtained in the early 1970s by academic researchers using laborious methods based on two-dimensional chromatography. Following the development of dye-based sequencing methods with automated analysis,[1] DNA sequencing has become easier and orders of magnitude faster.[2] History RNA sequencing was one of the earliest forms of nucleotide sequencing. The major landmark of RNA sequencing is the sequence of the first complete gene and the complete genome of Bacteriophage MS2, identified and published by Walter Fiers and his coworkers at the University of Ghent (Ghent, Belgium), between 1972[3] and 1976.[4] Prior to the development of rapid DNA sequencing methods in the early 1970s by Frederick Sanger at the University of Cambridge, in England and Walter Gilbert and Allan Maxam at Harvard,[5][6] a number of laborious methods were used. For instance, in 1973, Gilbert and Maxam reported the sequence of 24 basepairs using a method known as wandering-spot analysis.[7] The chain-termination method developed by Sanger and coworkers in 1975 soon became the method of choice, owing to its relative ease and reliability.[8][9] Maxam–Gilbert sequencing In 1976–1977, Allan Maxam and Walter Gilbert developed a DNA sequencing method based on chemical modification of DNA and subsequent cleavage at specific bases.[5] Although Maxam and Gilbert published their chemical sequencing method two years after the ground-breaking paper of Sanger and Coulson on plus-minus sequencing,[8][10] Maxam–Gilbert sequencing rapidly became more popular, since purified DNA could be used directly, while the initial Sanger method required that each read start be cloned for production of single-stranded DNA. However, with the improvement of the chain-termination method (see below), Maxam-Gilbert sequencing has fallen out of favour due to its technical complexity prohibiting its use in standard molecular biology kits, extensive use of hazardous chemicals, and difficulties with scale-up. The method requires radioactive labelling at one end and purification of the DNA fragment to be sequenced. Chemical treatment generates breaks at a small proportion of one or two of the four nucleotide bases in each of four reactions (G, A+G, C, C+T). Thus a series of labelled fragments is generated, from the radiolabelled end to the first "cut" site in each molecule. The fragments in the four reactions are arranged side by side in gel electrophoresis for size separation. To visualize the fragments, the gel is exposed to X-ray film for autoradiography, yielding a series of dark bands each corresponding to a radiolabelled DNA fragment, from which the sequence may be inferred. Also sometimes known as "chemical sequencing", this method originated in the study of DNA-protein interactions (footprinting), nucleic acid structure and epigenetic modifications to DNA, and within these it still has important applications. Chain-termination methods Because the chain-terminator method (or Sanger method after its developer Frederick Sanger) is more efficient and uses fewer toxic chemicals and lower amounts of radioactivity than the method of Maxam and Gilbert, it rapidly became the method of choice. The key principle of the Sanger method was the use of dideoxynucleotide triphosphates (ddNTPs) as DNA chain terminators. The classical chain-termination method requires a single-stranded DNA template, a DNA primer, a DNA polymerase, radioactively or fluorescently labeled nucleotides, and modified nucleotides that terminate DNA strand elongation. The DNA sample is divided into four separate sequencing reactions, containing all four of the standard deoxynucleotides (dATP, dGTP, dCTP and dTTP) and the DNA polymerase. To each reaction is added only one of the four dideoxynucleotides (ddATP, ddGTP, ddCTP, or ddTTP) which are the chain-terminating nucleotides, lacking a 3'-OH group required for the formation of a phosphodiester bond between two nucleotides, thus terminating DNA strand extension and resulting in DNA fragments of varying length. The newly synthesized and labeled DNA fragments are heat denatured, and separated by size (with a resolution of just one nucleotide) by gel electrophoresis on a denaturing polyacrylamide-urea gel with each of the four reactions run in one of four individual lanes (lanes A, T, G, C); the DNA bands are then visualized by autoradiography or UV light, and the DNA sequence can be directly read off the X-ray film or gel image. In the image on the right, X-ray film was exposed to the gel, and the dark bands correspond to DNA fragments of different lengths. A dark band in a lane indicates a DNA fragment that is the result of chain termination after incorporation of a dideoxynucleotide (ddATP, ddGTP, ddCTP, or ddTTP). The relative positions of the different bands among the four lanes are then used to read (from bottom to top) the DNA sequence.
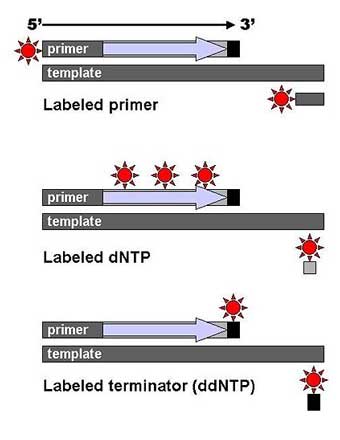
DNA fragments are labeled with a radioactive or fluorescent tag on the primer (1), in the new DNA strand with a labeled dNTP, or with a labeled ddNTP. Technical variations of chain-termination sequencing include tagging with nucleotides containing radioactive phosphorus for radiolabelling, or using a primer labeled at the 5’ end with a fluorescent dye. Dye-primer sequencing facilitates reading in an optical system for faster and more economical analysis and automation. The later development by Leroy Hood and coworkers [11][12] of fluorescently labeled ddNTPs and primers set the stage for automated, high-throughput DNA sequencing.
Sequence ladder by radioactive sequencing compared to fluorescent peaks (*) Chain-termination methods have greatly simplified DNA sequencing. For example, chain-termination-based kits are commercially available that contain the reagents needed for sequencing, pre-aliquoted and ready to use. Limitations include non-specific binding of the primer to the DNA, affecting accurate read-out of the DNA sequence, and DNA secondary structures affecting the fidelity of the sequence. Dye-terminator sequencing
Dye-terminator sequencing utilizes labelling of the chain terminator ddNTPs, which permits sequencing in a single reaction, rather than four reactions as in the labelled-primer method. In dye-terminator sequencing, each of the four dideoxynucleotide chain terminators is labelled with fluorescent dyes, each of which with different wavelengths of fluorescence and emission. Owing to its greater expediency and speed, dye-terminator sequencing is now the mainstay in automated sequencing. Its limitations include dye effects due to differences in the incorporation of the dye-labelled chain terminators into the DNA fragment, resulting in unequal peak heights and shapes in the electronic DNA sequence trace chromatogram after capillary electrophoresis (see figure to the left). This problem has been addressed with the use of modified DNA polymerase enzyme systems and dyes that minimize incorporation variability, as well as methods for eliminating "dye blobs". The dye-terminator sequencing method, along with automated high-throughput DNA sequence analyzers, is now being used for the vast majority of sequencing projects. Challenges Common challenges of DNA sequencing include poor quality in the first 15–40 bases of the sequence and deteriorating quality of sequencing traces after 700–900 bases. Base calling software typically gives an estimate of quality to aid in quality trimming. In cases where DNA fragments are cloned before sequencing, the resulting sequence may contain parts of the cloning vector. In contrast, PCR-based cloning and emerging sequencing technologies based on pyrosequencing often avoid using cloning vectors. Recently, one-step Sanger sequencing (combined amplification and sequencing) methods such as Ampliseq and SeqSharp have been developed that allow rapid sequencing of target genes without cloning or prior amplification. [13], [14] Current methods can directly sequence only relatively short (300–1000 nucleotides long) DNA fragments in a single reaction.[15] The main obstacle to sequencing DNA fragments above this size limit is insufficient power of separation for resolving large DNA fragments that differ in length by only one nucleotide. Automation and sample preparation
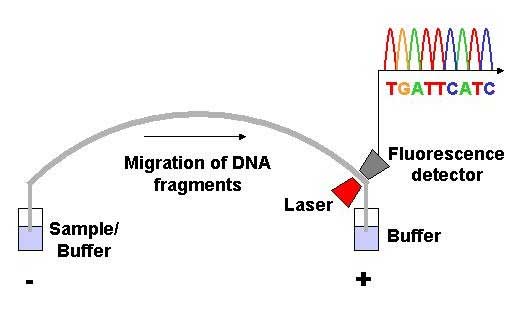
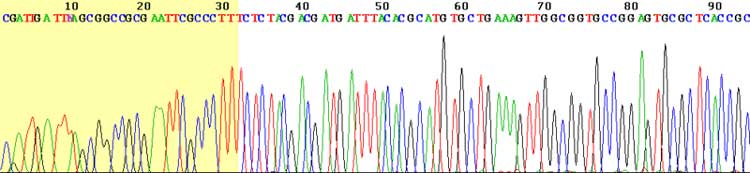
View of the start of an example dye-terminator read Automated DNA-sequencing instruments (DNA sequencers) can sequence up to 384 DNA samples in a single batch (run) in up to 24 runs a day. DNA sequencers carry out capillary electrophoresis for size separation, detection and recording of dye fluorescence, and data output as fluorescent peak trace chromatograms. Sequencing reactions by thermocycling, cleanup and re-suspension in a buffer solution before loading onto the sequencer are performed separately. A number of commercial and non-commercial software packages can trim low-quality DNA traces automatically. These programs score the quality of each peak and remove low-quality base peaks (generally located at the ends of the sequence). The accuracy of such algorithms is below visual examination by a human operator, but sufficient for automated processing of large sequence data sets. Amplification and clonal selection
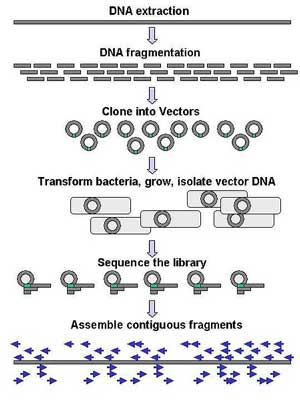
Genomic DNA is fragmented into random pieces and cloned as a bacterial library. DNA from individual bacterial clones is sequenced and the sequence is assembled by using overlapping DNA regions. Large-scale sequencing aims at sequencing very long DNA pieces, such as whole chromosomes. Common approaches consist of cutting (with restriction enzymes) or shearing (with mechanical forces) large DNA fragments into shorter DNA fragments. The fragmented DNA is cloned into a DNA vector, and amplified in Escherichia coli. Short DNA fragments purified from individual bacterial colonies are individually sequenced and assembled electronically into one long, contiguous sequence. This method does not require any pre-existing information about the sequence of the DNA and is referred to as de novo sequencing. Gaps in the assembled sequence may be filled by primer walking. The different strategies have different tradeoffs in speed and accuracy; shotgun methods are often used for sequencing large genomes, but its assembly is complex and difficult, particularly with sequence repeats often causing gaps in genome assembly. Most sequencing approaches use an in vitro cloning step to amplify individual DNA molecules, because their molecular detection methods are not sensitive enough for single molecule sequencing. Emulsion PCR[16] isolates individual DNA molecules along with primer-coated beads in aqueous droplets within an oil phase. Polymerase chain reaction (PCR) then coats each bead with clonal copies of the DNA molecule followed by immobilization for later sequencing. Emulsion PCR is used in the methods by Marguilis et al. (commercialized by 454 Life Sciences), Shendure and Porreca et al. (also known as "Polony sequencing") and SOLiD sequencing, (developed by Agencourt, now Applied Biosystems).[17][18][19] Another method for in vitro clonal amplification is bridge PCR, where fragments are amplified upon primers attached to a solid surface, used in the Illumina Genome Analyzer. The single-molecule method developed by Stephen Quake's laboratory (later commercialized by Helicos) is an exception: it uses bright fluorophores and laser excitation to detect pyrosequencing events from individual DNA molecules fixed to a surface, eliminating the need for molecular amplification.[20] High-throughput sequencing The high demand for low-cost sequencing has driven the development of high-throughput sequencing technologies that parallelize the sequencing process, producing thousands or millions of sequences at once.[21][22] High-throughput sequencing technologies are intended to lower the cost of DNA sequencing beyond what is possible with standard dye-terminator methods.[23] 454 pyrosequencing 454 Life Sciences has developed a parallelized version of pyrosequencing. It amplifies DNA inside water bubbles in an oil solution, each bubble containing a single initial DNA molecule and a single primer-coated bead that the DNA can attach to and form a clonal colony (emulsion PCR). The sequencing machine contains a large number of picolitre-volume wells that are large enough for a single bead, together with enzymes needed for sequencing. Pyrosequencing uses luciferase to generate light as read-out, and the sequencing machine takes a picture of the wells for every added nucleotide.[17] This technology provices intermediary read length and price per base compared to Sanger sequencing on one end and Solexa and SOLiD on the other.[24] Solexa sequencing Solexa has developed a sequencing technology based on reversible dye-terminators. DNA molecules are first attached to a primers on a slide and amplified so that local clonal colonies are formed (bridge amplification). One type of nucleotide at a time is then added, and non-incorporated nucleotides are washed away. Unlike pyrosequecning, the DNA can only be extended one nucleotide at a time. A camera takes images of the fluorescently labelled nucleotides and the dye is chemically removed from the DNA, allowing a next cycle.[25] SOLiD sequencing Applied Biosystems' SOLiD technology employs sequencing by ligation. Here, a pool of all possible oligonucleotides of a fixed length are labeled according to the sequenced position. Oligonucleotides are annealed and ligated; the preferential ligation by DNA ligase for matching sequences results in a signal informative of the nucleotide at that position. Before sequencing, the DNA is amplified by emulsion PCR. The resulting bead, each containing a only copies of the same DNA, are deposited on a glass slide.[26] Similar to Solexa sequencing, this technology produces short read lengths at a low price per base.[24] Future methods Sequencing by hybridization is a non-enzymatic method that uses a DNA microarray. A single pool of DNA whose sequence is to be determined is fluorescently labeled and hybridized to an array containing known sequences. Strong hybridization signals from a given spot on the array identifies its sequence in the DNA being sequenced.[27] Mass spectrometry may be used to determine mass differences between DNA fragments produced in chain-termination reactions.[28] DNA sequencing methods currently under development include labeling the DNA polymerase,[29] reading the sequence as a DNA strand transits through nanopores,[30][31] and microscopy-based techniques, such as AFM or electron microscopy that are used to identify the positions of individual nucleotides within long DNA fragments (>5,000 bp) by nucleotide labeling with heavier elements (e.g., halogens) for visual detection and recording.[32] In microfluidic Sanger sequencing the entire thermocycling amplification of DNA fragments as well as their separation by electrophoresis is done on a single glass wafer (approximately 10 cm in diameter) thus reducing the reagent usage as well as cost. In some instances researchers have shown that they can increase the throughput of conventional sequencing through the use of microchips. Research will still need to be done in order to make this use of technology effective. In October 2006, the X Prize Foundation established an initiative to promote the development of full genome sequencing technologies, called the Archon X Prize, intending to award $10 million to "the first Team that can build a device and use it to sequence 100 human genomes within 10 days or less, with an accuracy of no more than one error in every 100,000 bases sequenced, with sequences accurately covering at least 98% of the genome, and at a recurring cost of no more than $10,000 (US) per genome."[33] Major landmarks in DNA sequencing * 1953 Discovery of the structure of the DNA double helix.[34] * 1972 Development of recombinant DNA technology, which permits isolation of defined fragments of DNA; prior to this, the only accessible samples for sequencing were from bacteriophage or virus DNA. * 1977 The first complete DNA genome to be sequenced is that of bacteriophage φX174.[35] * 1977 Allan Maxam and Walter Gilbert publish "DNA sequencing by chemical degradation".[5] Frederick Sanger, independently, publishes "DNA sequencing with chain-terminating inhibitors".[36] * 1984 Medical Research Council scientists decipher the complete DNA sequence of the Epstein-Barr virus, 170 kb. * 1986 Leroy E. Hood's laboratory at the California Institute of Technology and Smith announce the first semi-automated DNA sequencing machine. * 1987 Applied Biosystems markets first automated sequencing machine, the model ABI 370. * 1990 The U.S. National Institutes of Health (NIH) begins large-scale sequencing trials on Mycoplasma capricolum, Escherichia coli, Caenorhabditis elegans, and Saccharomyces cerevisiae (at US$0.75/base). * 1991 Sequencing of human expressed sequence tags begins in Craig Venter's lab, an attempt to capture the coding fraction of the human genome.[37] * 1995 Craig Venter, Hamilton Smith, and colleagues at The Institute for Genomic Research (TIGR) publish the first complete genome of a free-living organism, the bacterium Haemophilus influenzae. The circular chromosome contains 1,830,137 bases and its publication in the journal Science[38] marks the first use of whole-genome shotgun sequencing, eliminating the need for initial mapping efforts. * 1996 Pål Nyrén and his student Mostafa Ronaghi at the Royal Institute of Technology in Stockholm publish their method of pyrosequencing[39] * 1998 Phil Green and Brent Ewing of the University of Washington publish “phred” for sequencer data analysis.[40] * 2001 A draft sequence of the human genome is published.[41][42] * 2004 454 Life Sciences markets a parallellized version of pyrosequencing.[43][44] The first version of their machine reduced sequencing costs 6-fold compared to automated Sanger sequencing, and was the first of a new generation of sequencing technologies.[24]
* Full genome sequencing
1. ^ Olsvik O, Wahlberg J, Petterson B, et al. (January 1993). "Use of automated sequencing of polymerase chain reaction-generated amplicons to identify three types of cholera toxin subunit B in Vibrio cholerae O1 strains". J. Clin. Microbiol. 31 (1): 22–5. PMID 7678018. PMC 262614. http://jcm.asm.org/cgi/pmidlookup?view=long&pmid=7678018.
* Disruptive Gene Sequencing technology - Single Molecule Real Time (SMRT) sequencing
Retrieved from "http://en.wikipedia.org/"
|

{kind=link}